Электронейромиография и ДНК-диагностика как основа современного медико-генетического консультирования при наследственной моторно-сенсорной невропатии (клиническое наблюдение)
ID: 2014-04-23-A-3901
Оригинальная статья (свободная структура)
ГБОУ ВПО Саратовский ГМУ им. В.И. Разумовского Минздрава России, кафедра неврологии ФПК и ППС им. К.Н. Третьякова
Резюме
.
Ключевые слова
наследственная моторно-сенсорная невропатия, электронейромиография, ДНК-диагностика
Статья
Научный руководитель: д.м.н., доцент Колоколов О.В.
Введение. Болезнь Шарко-Мари-Тута (невральная амиотрофия Шарко-Мари, наследственная моторно-сенсорная невропатия (НМСН)) объединяет группу генетически гетерогенных заболеваний периферической нервной системы (ПНС), характеризующихся симптомами прогрессирующей полинейропатии с преимущественным поражением мышц дистальных отделов конечностей. НМСН является самым частым среди наследственных заболеваний ПНС. Распространенность её в различных популяциях варьирует от 10 до 40 случаев на 100 тысяч населения. В настоящее время известно более 40 локусов и 20 генов, мутации в которых ответственны за развитие фенотипа НМСН.
Диагностика НМСН основывается на клинических данных, результатах электронейромиографии (ЭНМГ), а также ДНК диагностики, позволяющей не только достоверно определить тип мутации, но и провести медико-генетическое консультирование.
Все НМСН по результатам ЭНМГ и морфологическим признакам делят на три основных типа: 1) демиелинизирующий (НМСН I), характеризующийся снижением скорости проведения импульса (СПИ) (СПИ по срединному нерву < 38 м/с), 2) аксональный вариант (НМСН II), характеризующийся нормальной или несколько сниженной СПИ (СПИ по срединному нерву > 38 м/с), 3) промежуточный вариант со СПИ по срединному нерву от 25 до 45 м/с. ЭНМГ исследование имеет важнейшее значение для последующей ДНК-диагностики, так как позволяет определить оптимальный алгоритм молекулярно-генетического обследования для каждого пациента с наименьшими затратами времени и средств.
Несмотря на достижения нейрогенетики в диагностике НМСН, выявление этого заболевания очень часто происходит уже на поздних стадиях, когда эффективность лечебных и реабилитационных мероприятий низкая. Трудности ранней клинической диагностики связаны с малосимптомным течением болезни, фенотипическим полиморфизмом и генетической гетерогенностью. В настоящее момент лечение НМСН носит симптоматический характер.
Наиболее важной задачей лечения является сохранение моторных функций. Для этого рекомендуют индивидуально подобранную физиотерапию и лечебную гимнастику, но учитывают, что больным следует избегать перенапряжения. Для исправления проблем, вызванных деформацией стоп, могут использоваться специальные крепления, которые помогают контролировать тыльное сгибание стопы и голени, нестабильность голеностопного сустава и, зачастую, обеспечивают лучшее чувство равновесия. Аномалии ходьбы могут быть исправлены путем использования разных типов подтяжек (ankle-foot orthoses). Ортопедическая обувь также является важным направлением коррекции походки. Больных должен наблюдать подиатр. По мнению ряда ортопедов, определенный успех имеет хирургическая коррекция деформации стоп.
Клинический пример. Под нашим наблюдением находится пациент А., 33 лет, который предъявлял жалобы на слабость в мышцах дистальных отделов конечностей, деформацию стоп, нарушение походки, затруднения при передвижении в темное время суток. Оказалось, что с детства у него имелась особенность строения стоп (высокий свод), однако никаких затруднений в связи с этим он не испытывал. С 1996г. (16 лет) после перенесенной травмы (вывих правого голеностопного сустава) стала беспокоить слабость и утомляемость в мышцах дистальных отделов ног, что в то время связали с последствиями травмы. Спустя 10 лет (с 2006г.) стали заметными выраженные деформации стоп, нарушилась походка. В 2010г. присоединилась слабость в руках, нарушение чувствительности в стопах, в связи с чем стал затруднятся при передвижении в темное время суток. Однако к неврологам пациент не обращался. В сентябре 2012г. с описанными выше жалобами обратился к ортопедам (ФГБУ «СарНИИТО» МЗ РФ), которые заподозрили НМСН. Диагноз был подтвержден неврологом после проведения ЭНМГ. С целью коррекции грубой деформации стоп в ФГБУ «СарНИИТО» МЗ РФ произведен артродез правого голеностопного сустава.
В настоящее время в неврологическом статусе определяется симметричный дистальный периферический тетрапарез, более выраженный в ногах; нарушение чувствительности по полиневритическому типу; деформация стоп по типу Фридрейха; нарушения координации по типу сенситивной атаксии. Вышеописанное обуславливает выраженные нарушения походки.
При стимуляционной ЭНМГ обнаружены признаки грубого демиелинизирующего поражения моторных и сенсорных волокон периферических нервных стволов конечностей (с резким снижением СПИ), подтверждающее диагноз НИСН.
На основании анализа клинических данных и результатов ЭНМГ диагностирована НМСН Шарко-Мари-Тута I типа.
При обращении пациента проведено медико-генетическое консультирование, в рамках которого обсуждался вопрос о риске рождения у больного А. ребенка, который может заболеть НМСН.
При анализе родословной семьи А. (рис.1) оказалось, что признаки НМСН имеются не только у пробанда, но и у дяди по линии матери. Проявления заболевания у других сибсов, равно как и у других родственников, включая родителей пробанда, отсутствовали, что затруднило определение типа наследования. При аутосомно-доминантном типе наследования вероятность рождения ребенка (любого пола), который заболеет НМСН, составляет 50%. При Х сцепленном доминантном типе наследования все дочери больного отца будут больны, а сыновья – здоровы.
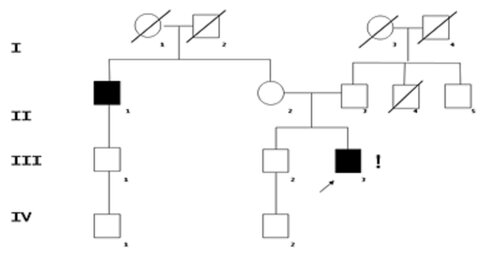
Рис. 1. Родословная
Молекулярно-генетические исследования проведены в лаборатории ДНК-диагностики МГНЦ РАМН. По результатам ДНК-анализа у пациента обнаружена наиболее частая при НМСН I типа мутация – дупликация на хромосоме 17p11.2-p12 (локус ШМТ1А). Таким образом, диагноз моторно-сенсорной нейропатии I типа окончательно подтвержден молекулярно-генетическими методами.
При проведении медико-генетического консультирования установлено, что ребенок, родившийся у пробанда, может заболеть НМСН с вероятностью 50 %.
Заключение. Особенностями данного клинического случая являются поздняя диагностика заболевания; быстро прогрессирующее течение с развитием деформации стоп и ранней инвалидизации, что потребовало проведения хирургической ортопедической коррекции. Кроме того, нетипичным является отсутствие признаков заболевания у родителей пробанда при аутосомно-доминантном типе наследования НМСН.
Выводы. Для обеспечения раннего выявления и профилактики инвалидизации при НМСН необходимо шире использовать методы молекулярно-генетической диагностики; повысить настороженность практикующих врачей в отношении данной патологии; обеспечить междисциплинарных подход в диагностике и ведении пациентов с НМСН, широко используя, как ортопедическую обувь и ортезы, так и (по строгим показаниям) методы хирургической коррекции деформации стоп.
Литература
- Гаусманова-Петрусевич И. Мышечные заболевания: Пер. с польского. - Варшава, 1971. - С. 370-384.
- Глущенко Е.В. Клинико-генетическая характеристика наследственной нейропатии Шарко-Мари-Тута: автореф. дис. … канд. мед. наук. – Красноярск, 2011.
- Евграфов О.В., Гроппа С.А. ДНК-диагностика наследственных заболеваний: Метод. рекомендации. - Кишинев, 1992. - 25 с.
- Лаукарт Е.Б. Клинико-диагностические аспекты деформаций стоп у неврологических больных и возможности реабилитационных мероприятий: автореф. дис. … канд. мед. наук. – М., 2011.
- Поповьян М.Д., Дубинская Е.Э. Эпидемиология и клиника наследственных болезней нервной системы. - Саратов, 1981. - 168 с.
- El-Abassi R., England J.D., Carter G.T. Charcot-Marie-Tooth Disease: An Overview of Genotypes, Phenotypes, and Clinical Management Strategies // PMR, 2014. Epub. 2014 Jan 13.
- Lee H.S. et al. Preimplantation genetic diagnosis for Charcot-Marie-Tooth disease // Clin Exp Reprod Med, 2013. Epub. 2013 Dec 31.
- Pelayo-Negro A.L. et al. Evolution of Charcot-Marie-Tooth disease type 1A duplication: a 2-year clinico-electrophysiological and lower-limb muscle MRI longitudinal study // J Neurol, 2014. Epub. 2014 Jan 22.
